

A research team led by Professor Yadong Wang from the School of Computing at Harbin Institute of Technology has made significant progress in bioinformatics algorithm development. They proposed a general-purpose cell abundance deconvolution framework for multi-omics data, achieving a transition from a “single-tool” approach to a “unified framework” paradigm, and establishing a new standard for integrative multi-omics analysis. The work, entitled “DECODE: A deep learning-based common deconvolution framework for various omics data”, has been published in Nature Methods.
Alongside the publication, the journal invited a team led by Robin Gasser from the Australian Academy of Science to write a News & Views article, providing a dedicated commentary highlighting the significance of this study.
Cellular composition and dynamic cell states are fundamental to understanding complex biological systems and disease progression. Omics data derived from tissue samples typically represent mixtures of signals from multiple cell types, making deconvolution algorithms essential for inferring cell-type proportions and states. In recent years, the rapid development of transcriptomics, proteomics, and metabolomics has led to the accumulation of large-scale cohort datasets. However, existing deconvolution methods are usually designed for a single omics modality and lack the ability to perform unified analysis across platforms, which greatly limits the integrative utilization of multi-omics data.
To address this challenge, the team developed DECODE, a general multi-omics deconvolution framework. This method is applicable to transcriptomic, proteomic, and metabolomic data simultaneously, enabling joint inference of cell types and cell states. Notably, it fills a longstanding gap in the field by providing the first generalizable framework for metabolomics deconvolution.
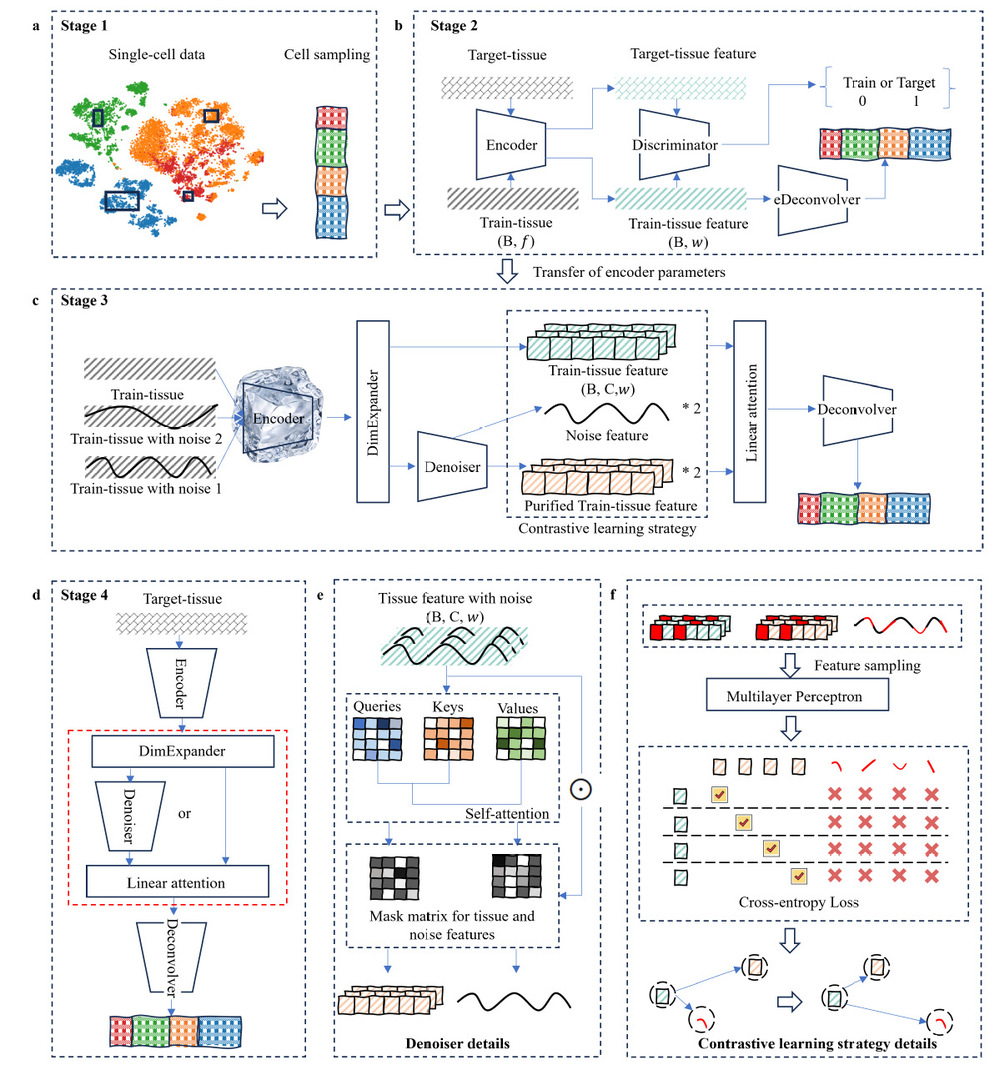
Schematic overview of the DECODE framework
The DECODE framework consists of several key modules. First, a simulation module is used to generate training data. Then, transfer adversarial learning is applied to align features across different omics modalities, effectively removing batch effects across platforms, disease states, and datasets. Furthermore, contrastive learning combined with self-attention mechanisms is employed to correct and denoise mixed signals in tissue samples, enabling accurate reconstruction of true cellular features. The synergy of these modules allows DECODE to robustly recover cell-type composition and cell states under complex data conditions.
Systematic benchmarking demonstrates that DECODE significantly outperforms state-of-the-art methods across different omics types, disease conditions, datasets, and measurement platforms. Importantly, it maintains high accuracy even when reference single-cell data are incomplete, highlighting its strong generalizability and robustness.
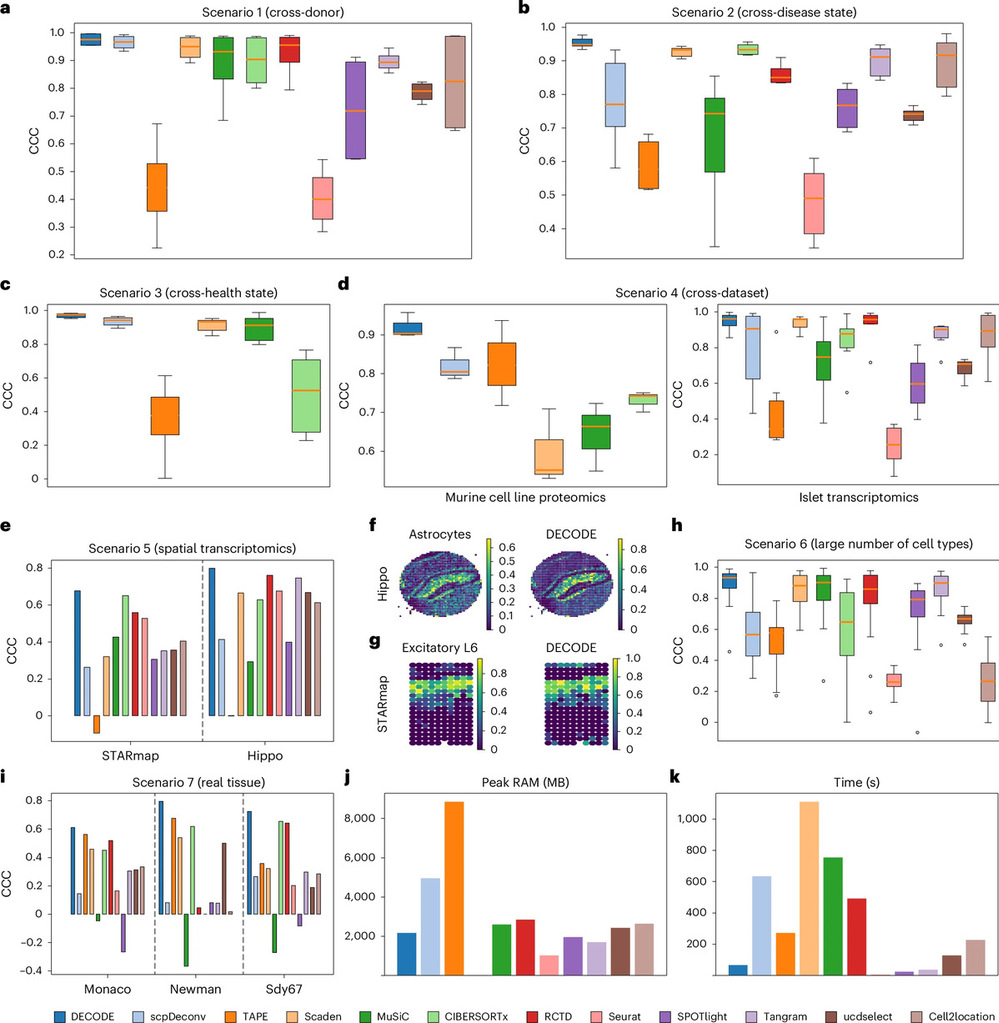
Performance of DECODE across different omics types, disease conditions, datasets, and measurement platforms
In the analysis of breast cancer multi-omics cohort data, DECODE revealed key changes in immune cell composition during tumor progression. For instance, in non-metastatic precancerous lesions, the proportion of T cells was significantly increased, while B cells were markedly decreased, consistent with previous findings that T cell infiltration is associated with favorable prognosis. Further subtype analysis showed that CD4⁺ T cells, CD8⁺ T cells, and proliferative T cells all increased during early stages, whereas the increase in B cells in late-stage tumors was mainly driven by naïve B cells, suggesting impaired immune function and enhanced metastatic potential.
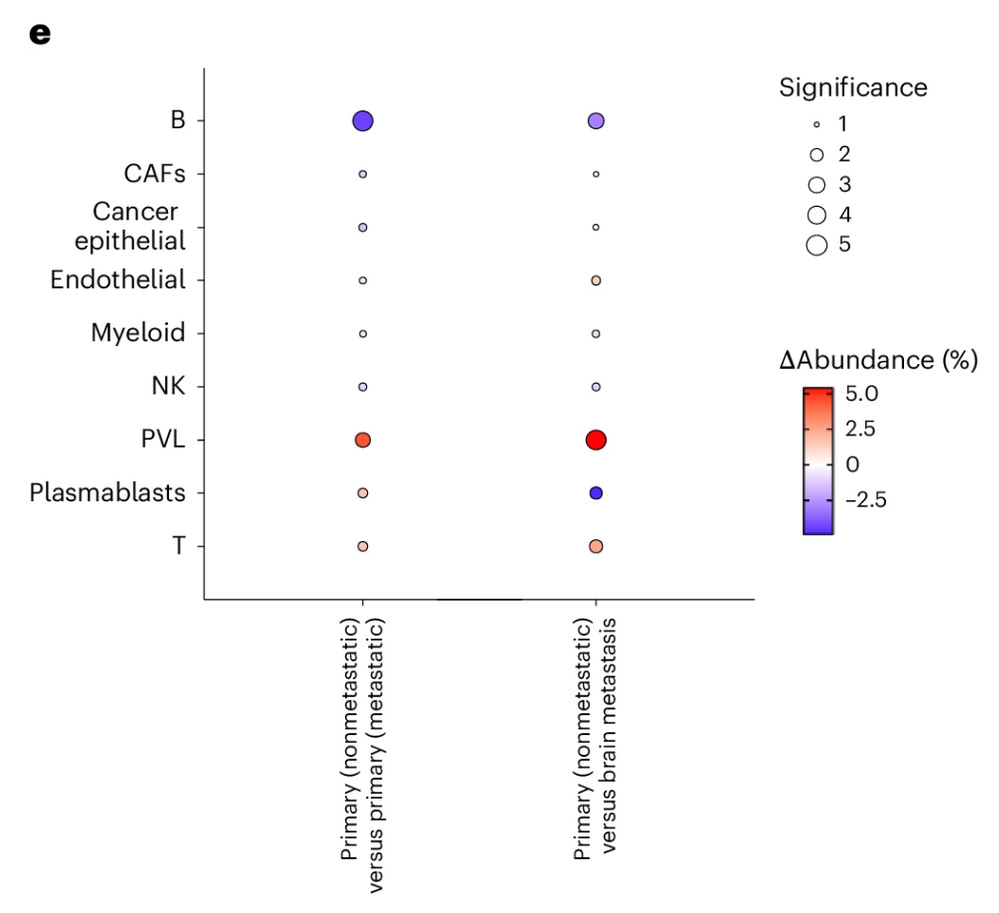
Results of DECODE applied to a breast cancer cohort dataset
This study presents a unified, cross-platform, and multi-omics deconvolution framework, filling a critical methodological gap and providing a powerful tool for mining large-scale multi-omics cohort data. DECODE is expected to serve as an important computational platform bridging basic research and clinical translation, offering new avenues for disease mechanism studies and precision medicine.
Harbin Institute of Technology is the first affiliated institution of this work. Professor Yadong Wang is the corresponding author. Professor Tianyi Zhao from the School of Life Science and Medicine, along with PhD students Renjie Liu and Yuzhi Sun from the School of Computing, are co-first authors. This research was supported by the National Natural Science Foundation of China and other funding sources.
Article link:
https://www.nature.com/articles/s41592-026-03007-y


